Buch-Navigation
Pigmente und Ablagerungen im Lymphknoten
DD braune/schwarze Pigmentierung:
- Anthrakose - schwarzes feingranuläres Pigment. Meist thorakale Lymphknoten, aber auch abdominell.
- Anthrakosilikose - schwarzes feingranuläres Pigment und Fibroseknötchen mit eingelagertem doppelbrechendem Fremdmaterial (Silikatkristalle). Meist thorakale Lymphknoten, aber auch abdominell.
- Tätowierpigment - Im Lymphabflussgebiet einer Tätowierung.
- Lipofuszin
- Hämosiderin
- Nodaler Nävus
- Melanommetastase!
- Dermatopathische Lymphadenopathie (s.u.)
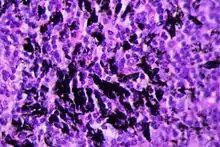 Anthrakosepigment im LK, H&E. |
Regressive Veränderungen
Fibrose (Hyalinose)
Lok.: Meist inguinal oder axillär.
Pg.: Ablagerung von kollagenem Bindegewebe.
SF.: Masson-Goldner, EVG.
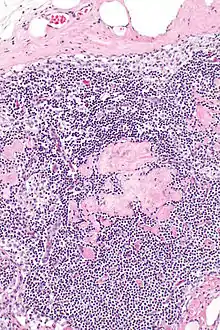 Hyalinose, H&E. |
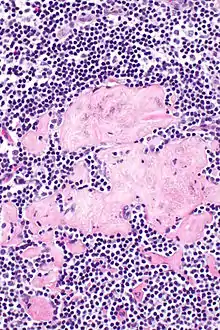 Idem. |
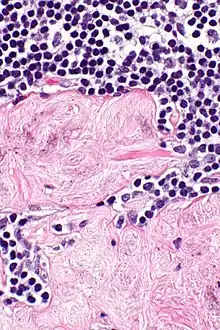 Idem. |
Lipomatöse Atrophie
Lok.: Meist inguinal oder axillär.
Pg.: Ersatz des lymphatischen Gewebes durch Fettgewebe.
%252C_HE_1.JPG.webp) Lipomatöse Atrophie, H&E. |
%252C_HE_2.JPG.webp) Idem. |
Verkalkung
Ät.: Postenzündlich, posttuberkulös.
Lok.: Oft mesenterial, intrathorakal.
Benigne Inklusionen
In Lymphknoten/Lymphofollikeln finden sich u.U. eingeschlossenene Gewebe benachbarter Organe. Sie sind eine wichtige Differentialdiagnose bei V.a. Lymphknotenmetastasen. Bsp.:
- Colonschleimhaut (im Bereich der Lymphfollikel ist die Lamina muscularis mucosae unterbrochen)
- Speicheldrüsengewebe in der Kopf-Hals-Region
- Schilddrüsengewebe in der Kopf-Hals-Region (DD: Metastase eines follikulären Schilddrüsenkarzinoms!)
- Bei Frauen:
- Endosalpingiose in der Beckenregion
- Endometriose
- Dezidua
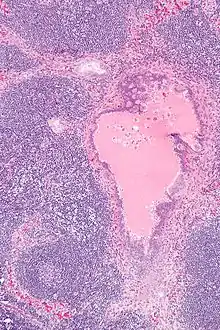 Endosalpingiose im Lymphknoten, H&E. |
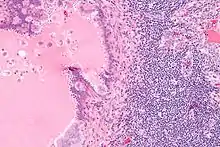 Idem. |
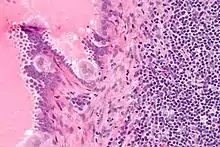 Idem. |
 Endometriose im Lymphknoten, H&E. |
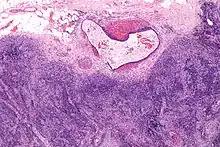 Idem. |
 Idem. |
 Dezidua im Lymphknoten, H&E. |
 Idem. |
 Idem. |
Reaktive Lymphknotenveränderungen
Mögliche Ursachen von Lymphknotenvergrößerungen:
- Infekte: Röteln, Masern, Windpocken, EBV (Pfeiffersches Drüsenfieber), Toxoplasmose, Tbc, Bartonella henselae (Katzenkratzkrankheit), HIV, Pseudo-Tbc (Yersiniose)
- Autoimmunerkrankungen: Sarkoidose
- Tumoren: Maligne Lymphome, benigne Lymphome (angiofollikuläre Lymphknotenhyperplasie - Morbus CASTLEMAN), Metastasen
- Dermatopathische Lymphadenopathie
Spezifisch vs. unspezifisch:
- Zur unspezifische Lymphadenitis gehören Sinushistiozytose, bunte Pulpahyperplasie und follikuläre Hyperplasie.
- Zeichen der spezifischen Lymphadenitis sind z.B. Epitheloidzellgranulome.
Histologische Typen:
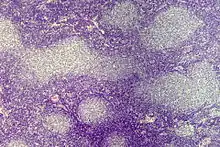
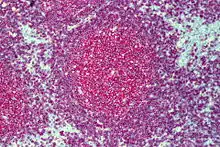
Lymphofollikuläre Hyperplasie
Makro: Lymphknoten (oder enstpr. lymphatisches Gewebe) oft vergrößert.
Histo: Zahlreiche große scharf begrenzte Sekundärfollikel mit großen Keimzentren mit heller und dunkler Zone sowie Sternenhimmelbild durch eingestreute große, helle Kerntrümmer-Makrophagen (tingible-bodies-Makrophagen), im Lymphknoten zonale Gliederung (Rinde, Parakortex, Mark).
DD: Malignes follikuläres Lymphom: Unscharf begrenzte Follikel, kein zonales Phänomen, monoton, kein Sternenhimmel.
Gegenüberstellung:
| Kriterium | Reaktive follikuläre Hyperplasie | Follikuläres Lymphom | ||
|---|---|---|---|---|
| Follikel | unterschiedlich groß, ungleichförmig | 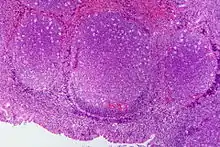 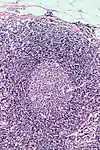 | 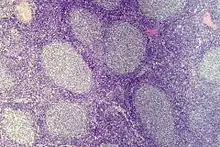 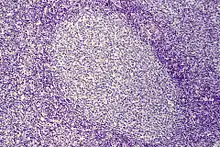 | gleich groß, gleichförmig |
| Kerntrümmer-Makrophagen | ja | nein | ||
| Polarität | ja | nein | ||
| Zonale Gliederung | ja | vermindert oder fehlend | ||
| Extranodale Ausbreitung | nein | ja | ||
| Proliferation (MIB-1) | hoch | 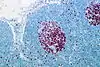 | 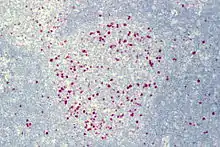 | niedrig |
| bcl-2 im Keimzentrum | nein | 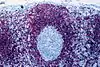 | 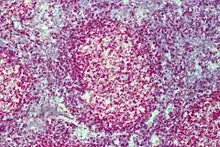 | meist ja |
| CD 10 | negativ |  | 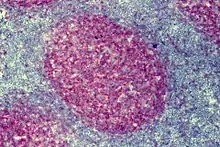 | häufig positiv |
Progressive Transformation der Keimzentren (PTGC)
progressive transformation of germinal centers
Ät.: unklar.
Histo:
- Follikuläre Hyperplasie (s.o.)
- Große Follikel mit verbreiterter Mantelzone und unscharfer Keimzentrum/Mantelzone-Grenze.
Klinik: Lokale schmerzlose Lymphknotenschwellung.
PTGC, H&E. |
Idem. |
Idem. |
Idem. |
Idem. |
Interfollikuläre Hyperplasie
Ät.: Rheumatische Erkrankungen
Mikro: Breite interfollikuläre Zone und bunte Pulpahyperplasie (T-Lymphozyten, Plasmazellen, eingestreute kleine Blasten).
Sinusoidale Hyperplasie (Sinushistiozytose)
Unspezifische reaktive benigne Veränderung mit Vermehrung der Histiozyten in den Sinus.
Ep.: Häufig.
Ät.: Entzündungen, Tumor, Anthrakose, Rauchen.
Histo: Follikel scharf begrenzt, parafollikuläre Zone, Histiozyten (helle Zellen) in den Sinusoiden stark vermehrt. (Cave: Kann Lymphknoten-Metastasen sehr ähneln).
IHC: CD68 +, CD1a -, S100 - (Follikulär-dendritische Zellen, FDC +)
DD:
- Karzinom/Melanom-Metastase - CD68 -, Karzinom CK +, Melanom Melan A +, HMB45 +
- LANGERHANS-Zell-Histiozytose - CD1a +
- ROSAI-DORFMAN-Erkrankung - S100 +
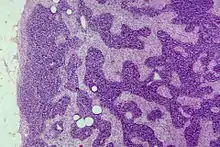 Sinushistiozytose, H&E. |
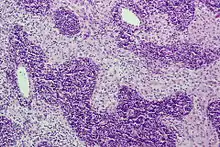 Idem. |
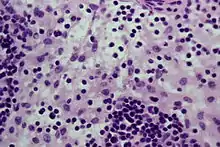 Idem. |
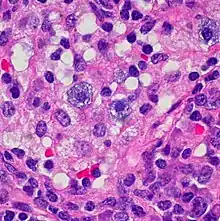 Vermehrung der Mastzellen bei Sinushistiozytose. |
Nekrotisierend-abszedierende Lymphadenitis
Ät.: Katzenkratzkrankheit (Bartonella henselae), Pest (Yersina pestis), Tularämie (Francisella tularensis ), Lymphogranuloma venereum (Chlamydia trachomatis).
Histo:
- Lymphknoten mit Nekrosen
- Abszedierung (neutrophile Granulozyten).
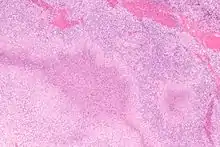 Katzenkratzkrankheit, H&E. |
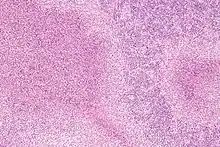 Idem. |
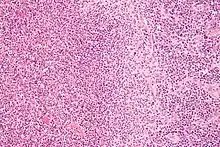 Idem. |
Nekrotisierende Lymphadenitis bei SLE
Histo:
- Nekrosen mit Hämatoxilin-bodies.
- Keine neutrophile Infiltration.
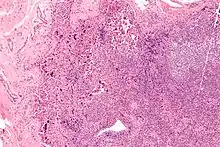 Lymphadenitis bei SLE, H&E. |
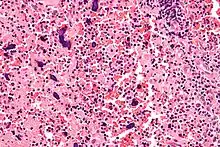 Idem. |
Nekrotisierende histiozytäre Lymphadenitis (KKIKUCHI-Erkrankung)
Ep.: Junge Erwachsene. Selten.
Ät.: Unklar.
Lok.: Zervikal.
Prg.: Selbstlimitierend.
DD.: Morbus HODGKIN, Katzenkratzkrankheit, LK-Tuberkulose.
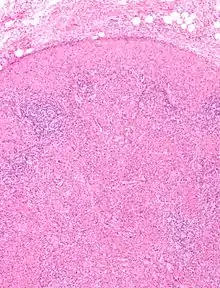 Nekrotisierende histiozytäre Lymphadenitis, H&E. |
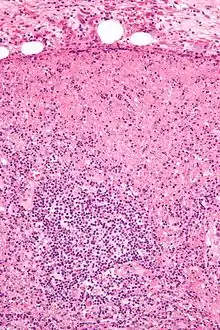 Idem. |
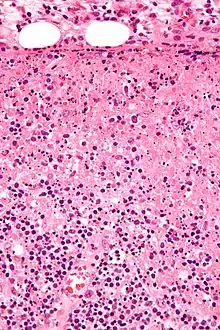 Idem. |
Granulomatöse Lymphadenitis
Granulom: Klein- oder großherdige Ansammlung von Histiozyten, Riesenzellen, Epitheloidzellen und Lymphozyten mit oder ohne Nekrose.
- Kleine Granulome: Toxoplasmose
- Große Granulome: Sarkoidose, Tbc, Pseudo-Tbc
Andere: Reaktiv, Lues Stadium III, Brucellose, viszerale Leishmaniose, Lepra, Schistosomiasis, Morbus CROHN, WEGENER-Granulomatose, Morbus WHIPPLE, Pilze.
Sarkoidose
Siehe auch Kapitel Untere Atemwege und Lungen
Histo:
- Granulome aus Epitheloidzellen und Histiozyten
- keine käsige Nekrose
- mehrkernige Riesenzellen.
Sarcoid-like lesion
Reaktive Sarkoidose-ähnliche Epitheloizellgranulome in Lymphknoten, die im Abflussgebiet von malignen Tumoren liegen.
Histo: Knotige Aggregate aus Epitheloidzellen (eosinophiles Zytoplasma, schlecht abgrenzbare Zellgrenzen, schuhsohlenartige Kerne, helles Karyoplasma, kleine Nukleolen).
Lit.:
- Ravaglia C, Gurioli C, Casoni GL, et al.. “Sarcoid-like lesion is a frequent benign cause of lymphadenopathy in neoplastic patients”. Eur. Respir. J., 41:754–5, March 2013. DOI:10.1183/09031936.00141212. PMID 23456935.
- GREGORIE HB, OTHERSEN HB, MOORE MP. “The significance of sarcoid-like lesions in association with malignant neoplasms”. Am. J. Surg., 104:577–86, October 1962. PMID 13901642.
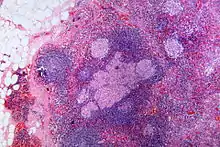 Sarcoid-like lesion im Abflussgebiet eines oropharyngealen Plattenepithelkarzinoms, H&E. |
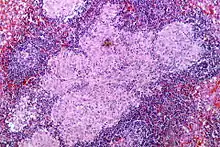 Idem. |
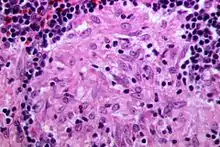 Idem. |
Morbus CROHN
Siehe Kapitel Intestinum tenue und Colon.
Histo: Nicht-verkäsende Granulome, ggf. mit Riesenzellen (u.U. vom LANGHANS-Typ).
 Morbus Crohn, intestinaler Lymphknoten, H&E. |
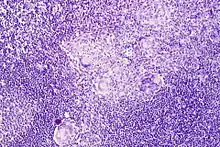 Idem. |
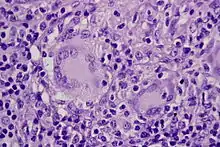 Idem. |
Toxoplasmose
Ät.: Toxoplasma gondii.
Histo:
- Epitheloidzellaggregate.
- Aggregate aus monozytoiden B-Zellen.
- Unscharf begrenzte reaktive Keimzentren.
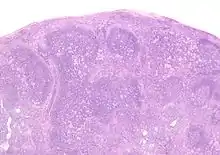 Toxoplasmose, H&E. |
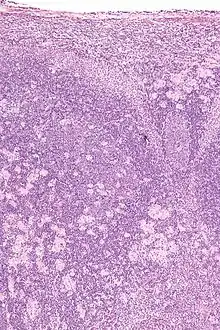 Idem. |
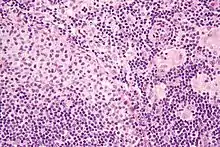 Idem. |
Atypische Mykobakteriosen
RF.: Infektion mit atypischen Mykobakterien (MOTT) bei Immunsuppression.
Histo: Granulomatöse Entzündung.
SF: ZIEHL-NEELSEN.
_1.jpg.webp) Atypische Mykobakteriose, H&E. |
_2.jpg.webp) Idem. |
_3.jpg.webp) Idem. |
_4.jpg.webp) Idem, AFB stain. |
Dermatopathische Lymphadenopathie
Ät.: Mit Hauterkrankungen assoziierte Lymphadenopathie.
Histo:
- Melanophagen.
- Sinushistiozytose.
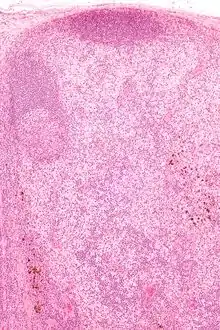 , H&E. |
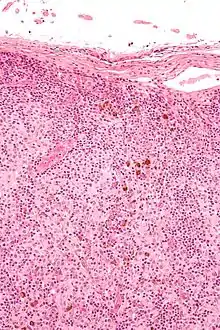 Idem. |
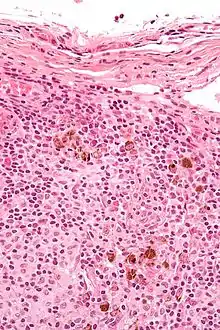 Idem. |
Lymphoproliferative Erkrankungen
Post-Transplant Lymphoproliferative Disorder (PTLD)
Syn.: Lymphoproliferative Erkrankung nach Transplantation.
Ät.: EBV-Infektion + immunsuppressive Therapie nach Organtransplantation.
Morbus CASTLEMAN
Syn.: Angiofollikuläre Hyperplasie.
Ät.: Die Plasmazellreiche Variante ist assoziiert mit HIV und HHV8.
Histo:
- Hyalin-vaskuläre Variante:
- Kleine regressive Keimzentren.
- Breite Mantelzone.
- Sklerosierte auf die Keimzentren ausgerichtete Gefäße („Lollipop“-Aspekt).
- Sinus-Verlust.
- Plasmazellreiche Variante.
Klinik:
- Lokalisiert: Asymptomatisch oder Allgemeinsymptome.
- Multizentrisch: Allgemeinsymptome, unspezifische Störungen (PNP, Hautveränderungen, hormonelle Störungen), Lymphome.
Th.:
- Lokalisierte Form: Chirugische Resektion.
Prg.: Bei der multifokalen Form ungünstig.
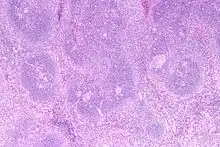 Morbus CASTLEMAN, H&E. |
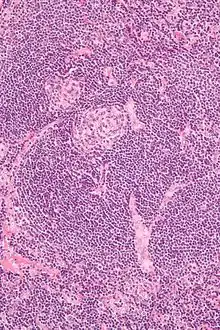 Idem. |
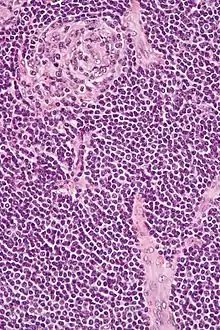 Idem. |
ROSAI-DORFMAN-Erkrankung
Syn.: Sinushistiozytose mit massiver Lymphadenopathie.
Ät.: unklar.
Histo:
- Histiozytenvermehrung
- Emperipolesis (Phagozytose)
IHC: S-100 +, CD1a -
Klinik: Lymphknotenschwellung, Allgemeinsymptome.
Prg.: Meist selbstlimitierend.
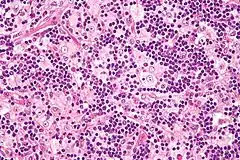 ROSAI-DORFMAN-Erkrankung, H&E. |
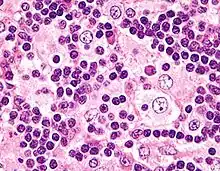 Idem. |
LANGERHANS-Zellen-Histiozytose
Syn.: Histiozytose X
Systemische oder lokalisierte Proliferation bzw. Tumor der LANGERHANS-Histiozyten.
Histo: Proliferation von LANGERHANSZellen (reniforme Kerne mit Kernfalten)
IHC: S-100 +, CD 1a +.
Mol.Path.: BRAF-Mutationen (V600E).
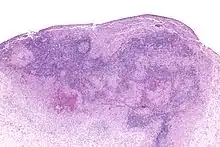 LANGERHANS-Zellen-Histiozytose, H&E. |
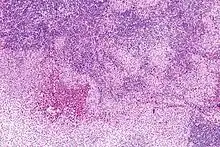 Idem. |
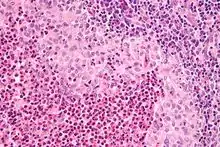 Idem. |
Morbus ABT-LETTER-SIWE
LANGERHANS-Histiozytose mit Beteiligung von Leber, Lunge und Knochenmark.
Ep.: Manifestation im 1. - 2. Lj.
Klinik: Fieber, Petechien, Knochenerweichung, Hepatosplenomegalie, generalisierte Lymphadenopathie, Lipoideinlagerungen im granulomatösen Gewebe.
Prg.: In 50% letaler Verlauf.
Morbus HAND-SCHÜLLER-CHRISTIAN
Cholesterinlipidose (Lipoidgranulomatose) mit Cholesterinspeicherung im Monozyten-Makrophagen-System und Histiozyten-Proliferation.
Ep.: Meist Kinder unter 5 Jahren.
Verhalten: Benigne.
Klinik: Lytische Knochenläsionen, Exophthalmus, Diabetes insipidus, Landkartenschädel
Eosinophiles Knochengranulom
Ansammlungen von teils mehrkernigen Langerhanszellen mit eosinophilen Granulozyten u.a. im Knochen.
Ep.: Junge Erwachsene.
Klinik: Scharf begrenzte osteolytische Herde v.a. im Schädel, proximalen Femur, Becken, Wirbelsäule.
Prg.: Rückbildung spontan oder nach Curretage.
_jaw_bone.jpg.webp) Eosinophiles Knochengranulom des Kiefers, Knochenbiopsie, H&E. |
_jaw_bone.jpg.webp) Idem. |
_jaw_bone.jpg.webp) Idem. |
_jaw_bone.jpg.webp) Idem. |
_S-100.JPG.webp) Idem, S-100-Immunfärbung. |
Maligne Lymphome
Lymphopoese
- Knochenmarkstammzelle -> Prä-T-Zelle -> T1-Lymphozyt -> T-Immunoblast -> T2-Lymphozyt
- Knochenmarkstammzelle -> Prä-B-Zelle -> B1-Lymphozyt
- B1-Lymphozyt -> B-Immunoblast -> Lymphoplasmozytoide Zelle -> Plasmazelle
- B1-Lymphozyt -> Zentroblast -> B-Immunoblast -> s.o.
- B1-Lymphozyt -> Zentroblast -> Zentrozyt -> B-Immunoblast -> s.o.
Histologie normales Knochenmark
- Knochenbälkchen (Jugend: mehr, Alter/Osteoporose: weniger)
- Markraum
- Fett (Alter: mehr Fett, Fett:Mark = ca. 3:2 mit 60 Jahren)
- Hämatopoetisches Mark
- Erythropoese - Erythroblasten rundlich, dunkel, kompakt in Haufen gelagert
- Granulopoese - zahlreich entlang der Knochenbälkchen
- Megakaryozytopose - Disseminierte große mehrkernige Zellen
Klassifizierung der Lymphome:
- Morbus HODGKIN - Non-HODGKIN-Lymphome (NHL)
- B-Zell - T-Zell
- Niedrigmaligne (indolent) - hochmaligne (aggressiv)
- Typ und Subtyp anhand Morphologie, Antigenexpression, Zytogenetik
Klinik: Schmerzlose Lymphknotenschwellung, B-Symptome (Nachtschweiß, Fieber, Gewichtsverlust), Zytopenie (Anämie, Thrombopenie, Leukopenie), Infekte, Kompressionssyndrome (z.B. Harnstauungsniere).
Morbus HODGKIN
Ep.: Mittleres Alter
Typen:
- Klassisch lymphozytenreich, nodulär-diffus (ca. 5 %)
- Noduläre Sklerose (ca. 80 %)
- Gemischte Zellularität (ca. 15 %)
- Lymphozytendepletion (selten)
Makro:
- Beginn in den Lymphknoten: Vergrößert, derb, verbacken
- Typisch: Mediastinaltumor
- Bauernwurstmilz (noduläre Infiltration)
- Leberbefall (Portalfelder)
- Knochenmarksbefall u.a.
Histo Lymphknoten: Bindegewebig abgetrennte Knoten, nodulär, zerstörte Lymphknotenarchitektur, eingestreute Tumorzellen in Form von HODGKIN-Zellen (Große Zellen mit großen hellen Kernen und dunklen Nukleolen, umgeben von einem Halo) und REED-STERNBERG-Zellen (HODGKIN-Zell-Pärchen, „Schielaugen“), viele reaktive Zellen, buntes gemischtzelliges Zellbild.
IHC: CD 30 +, CD 15 +
Klinische Klassifikation nach Ann Arbor:
- I.: Befall einer Lymphknotenstation
- II.: Befall mehrere Lymphknotenstationen auf einer Zwerchfellseite
- III.: Befall mehrere Lymphknotenstationen auf beiden Seiten des Zwerchfells
- IV.: Multipler Organbefall
.jpg.webp) Stark vergrößerte axilläre Lymphknoten bei Morbus HODGKIN. |
_mixed_cellulary_type.jpg.webp) Gemischt-zelluläres HODGKIN-Lymphom, Lymphknotenbiopsie, H&E. |
_mixed_cellulary_type.jpg.webp) Idem. |
_mixed_cellularity_type.jpg.webp) Idem. |
_CD30_immunostain.jpg.webp) Idem, IHC auf CD 30 (Ki-1). |
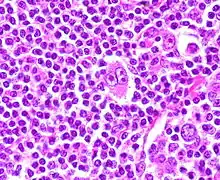 REED-STERNBERG-Zelle bei Morbus HODGKIN. Ein anderer Fall. |
B-Zell-Lymphome (NHL)
Typen:
- Vorläuferlymphoblastische Lymphome/Leukämien
- CLL (niedrigmaligne)
- Lymphoplasmozytäres Lymphom (Immunozytom, Morbus WALDENSTRÖM)
- Lymphknotenplasmozytom (klinisch hoch-, pathologisch niedrigmaligne keine Blasten)
- Follikuläre Lymphome I-III (I-II niedrigmaligne, später maligne (RICHTER-Syndrom!), zentroblastisch, zentrozytisch), ca. 30 %
- Mantelzell-Lymphom (zentrozytisch)
- Marginalzonen-Lymphom (MALT, splenisch, nodal), häufigstes Lymphom im Magen
- Haarzell-Leukämie
- Diffuses großzelliges B-Zell-Lymphom (zentroblastisch, immunoblastisch), ca. 30 %
- Burkitt-Lymphom (hochmaligne)
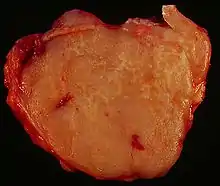 Malignes high-grade B-Zell-Lymphom. |
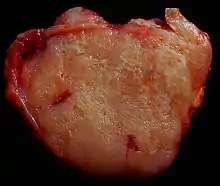 Idem, andere Beleuchtung. |
Diffus großzelliges B-Zell-Lymphom
Ep.: Häufigstes hoch malignes NHL.
Th.: R-CHOP (Rituximab, Cyclophosphamid, Hydroxydaunorubicin, Oncovin/Vincristin, Prednison), ggf. Radiatio, ggf. ablative Chemo + SZT.
Prg.: 50 – 70 % Heilungsrate
_tonsil.jpg.webp) Diffuses großzelliges B-Zell-Lymphom der Tonsille, H&E. |
_tonsil.jpg.webp) Idem, stärkere Vergrößerung, große helle Tumorzellen z.T. mit angedeuteten Radspeichen. |
_CD20.jpg.webp) Idem, Immunfärbung von CD 20 (B-Zellmarker). |
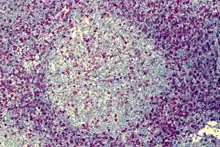
Follikuläres Lymphom
Ep.: Häufigstes niedrig malignes NHL. 6. Dekade.
Ursprung: Follikelzentrum B-Zellen (Zentrozyten and Zentroblasten).
Ät.: Häufig Translokation zwischen Chromosom 14 und 18 mit Überexpression des Antiapoptose-Faktors Bcl-2 (bcl-2, Chromosom 18) unter dem Einfluss des immunoglobulin heavy chain enhancer element (Chromosom 14) mit konsekutiver Immortalisierung der Zellen. Möglich sind auch Translokationen von bcl-6.
Zytogenetik: t(14;18)
Histo: Gleichförmige dicht gelagerte atypische Lymphfollikel mit monotonen apolaren Keimzentren (keine helle und dunkle Zone) ohne Sternhimmelmakrophagen und ohne zonale Gliederung der Lymphknoten (Rinde - Mark), reduzierte/fehlende Mantelzone. Extranodale Ausbreitung.
IHC: CD20 +, CD19 +, Bcl-2 +, evtl. Bcl-6 +, CD10 +, CD3 -, CD 5 -, Ki67/Mib-1 reduziert!.
Grading:
| Grad | Kriterium |
|---|---|
| 1 (low-grade) | 0 - 5 Zentroblasten pro HPF |
| 2 (low-grade) | 6 - 15 Zentroblasten pro HPF |
3 (high-grade)
|
> 15 Zentroblasten pro HPF
|
DD.: Reaktive lymphofollikuläre Hyperplasie!
Klinik: Oft oligosymptomatisch, indolente Lymphknotenschwellung.
Th: Bei Beschwerden wie Kompression (Ureter, Gefäße), Zytopenie, Infekte, B-Symptome. Z.B. R-CHOP.
Follikuläres Lymphom , H&E. |
 Idem. |
Idem. |
 CD20 + (B-Marker). |
CD10 +. |
 CD3, reaktive T-Lymphozyten. |
BCL2 +. |
Mib-1 dtl. reduziert im Vgl. zu reaktiven Keimzentren. |
Chronische lymphatische Leukämie (CLL)
Ep.: Häufigste Leukämie in westlichen Ländern, Alter > 40 Jahre, im Mittel 60 Jahre, nicht selten Zufallsbefund.
Subtypen:
- 95 % B-Zell, 5 % T-Zell.
- Small lymphocytic lymphoma (SLL) - Vorwiegend nodale aleukämische Verlaufsform.
Makro: Hepatosplenomegalie (Tumorinfiltrat). Befallene Lymphknoten sind vergrößert, markig-weich, weißlich.
Histo: Primär Infiltration des Knochenmarks durch lymphozytäre Elemente, sekundär Ausschwemmung ins Blut (Leukos 15.000 - 100.000).
IHC:
- B-Marker: CD19, CD20, CD79a.
- T-Marker: CD3, CD4, CD5, CD8.
- Proliferationsmarker: MIB1 niedrig (5 %).
- Weitere Marker: CD5 +, CD23 +, CD10 -, Cyclin D1 -.
- Leichtkettenrestriktion (kappa oder lambda).
Blutausstrich: Starke Vermehrung reifer unauffälliger Lymphozyten (Leukozytose, Lymphozytose), viele GUMPRECHT-Kernschatten (Ausstrich-Artefakte).
DD.:
- Mantelzelllymphom: Cyclin D +.
Verhalten: Niedrig maligne.
Klinik: Indolente Lymphknotenschwellungen, leichte Splenomegalie, B-Symptome (Fieber, Nachtschweiß, Gewichtsverlust), opportunistische Infektionen, Anämie, Thrombopenie, Granulozytopenie, Antikörpermangel-Syndrom. Klinische Stadieneinteilung nach BINET.
Kompl.: Hypogammaglobinämie, Infektionen (häufigste Todesursache bei CLL), RICHTER-Syndrom (Übergang in ein hochmalignes Lympom).
.jpg.webp) B-CLL mit typischen Ausstrichartefakten, Blutausstrich, Wright-Giemsa stain. |
|
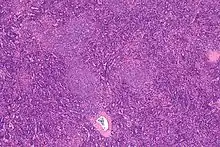
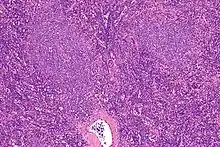
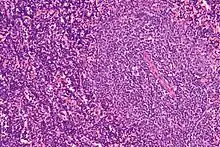
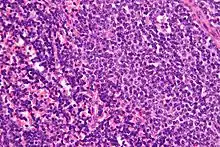
Haarzellleukämie
Hairy cell leukemia
SF der CLL
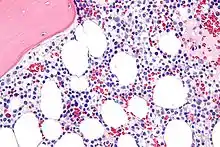 HCL, Knochenmark, H&E. |
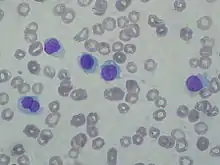 HCL, Blutausstrich. |
Mantelzelllymphom
Makro im Magen: Tumor submukös (DD: Karzinom), Ulkus, Faltenbildung fehlt.
Mikro: Kleine, monotone, blaue Zellen, zentrozytenartig. Lymphom ringförmig um den normalen Lymphfollikel angeordnet. Mitosen, sklerosierte Blutgefäße, Epitheloidzellen.
Immunhistochemie:
- CD 43 +, CD 45 (LCA) +, CD 23 -
- B: CD 20 +, CD 79a +
- T: CD 3 -, CD 5 +, CD 8 -
- Proliferationsmarker: MIB1 niedrig (~10 %) (Ausnahme blastenreicher Typ, hier MIB-1_LI von 50 - 60 %), CD 10 -, Cyclin D1 +.
Mol: Translokation t(11;14)(q13;q32) -> CCND1/IGHG1-Fusionsgen.
Pathologisch niedermaligne, klinisch hochmaligne.
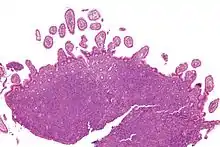 Mantelzelllymphom im Ileum, H&E. |
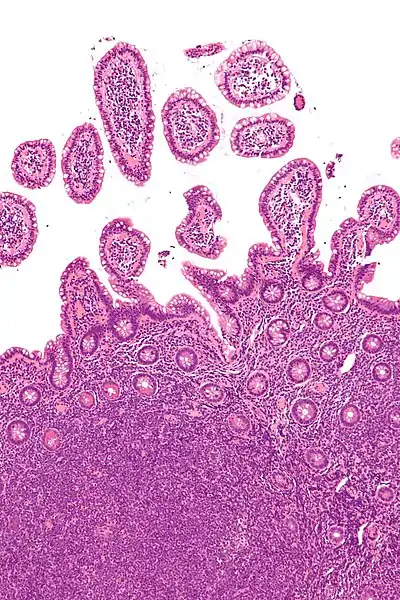 Idem. |
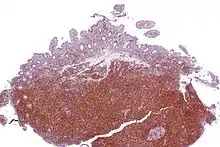 Idem, Cyclin D1. |
Plasmozytom
plasmacytoma
Ep.: Häufigster maligner Tumor des Knochens, 6.-7. Dekade, Dunkelhäutige 3x häufiger betroffen.
Ät.: Unklar, chronische Antigenstimulation, assoziiert mit HLA4c, genetisch-ethnische Faktoren, Z.b. Bestrahlung/Chemotherapie.
Formen:
- Solitäres Plasmozytom
- Multiples Myelom
- Extramedulläres Myelom der Weichteile
Produzierter monoklonaler Immunglobulin-Typ:
- IgG 65 %
- IgA 20 %
- BENCE-JONES-Plasmozytom: Nur leichte Ketten (meist kappa)
- Andere: selten
Makro:
- Knochenmarksverdrängung -> Anämie, Leukopenie, Thrombopenie
- Osteolysen (Tumor produziert Osteoklastenaktivierenden Faktor): ausgestanzte Defekte in Wirbelsäule, Schädel (Rö: Schrotschußschädel (eher selten)), Rippen.
- Extraossäre Infiltrate: periostal (paravertebral), evtl. Milz, Leber, Lymphknoten.
Histo:
- > als 10 % typische Plasmazellen mit exzentrischem großem rundem Kern, breitem mantelartigem Zytoplasmasaum und diskretem perinukleärem Halo aus hellerem Zytoplasma („Spiegeleier“), evtl. Atypien.
- RUSSEL-bodies (eosinophile globuläre Immunglobulin-deposits).
Immunhistochemie:
- B- und Plasmazell-Marker + : CD 79a +
- Je nach Typ Ig +, kappa/lambda-light-chains +
Weitere Folgen: Amyloidose (10 - 15 %), Hyperviskositätssyndrom, monoklonale Gammopathie -> Infekte, Gerinnungsfaktor-Antikörper, Plasmozytomniere (Plasmazellinfiltrate, Amyloid, Tubulusepithelschäden, Kalkablagerungen, chronische Pyelonephritis).
Klinik: Paraproteinämie (M-Gradient in der S-Elektrophorese), Zytopenien, BSG-Beschleunigung, Osteolysen, Infekte, Niereninsuffizienz.
Verhalten: Klinisch hochmalignes Lymphom, morphologisch nicht immer.
Klinische Stadieneinteilungen:
nach SALMON und DURIE:
| International Staging System (ISS) 2005:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Therapieoptionen:
- medikamentös:
- Chemotherapie
- Kortikosteroide (Prednison, Dexamethason)
- Bortezomib (Proteasominhibitor)
- Lenalidomid, Thalidomid
- Bisphosphonate
- autologe Stammzelltransplantation
- lokale Radiatio
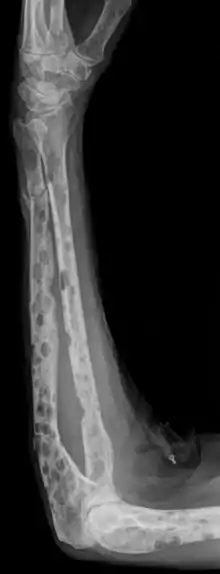 Muliple Osteolysen. |
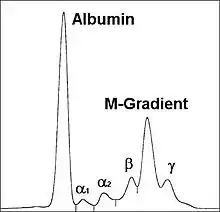 M-Gradient. |
_HE_stain.jpg.webp) Multiples Myelom, Knochenmarkaspirat, H&E. |
_MG_stain.jpg.webp) Idem, Ausstrichpräparat des Knochenmarkaspirats, gefärbt nach May-Grünwald-Giemsa. |
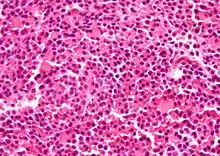 RUSSEL-bodies, H&E. |
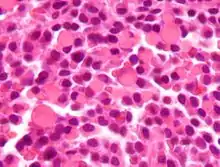 Idem. |
Extramedulläres Plasmozytom der Weichteile
Ep.: Selten.
Lok.: 75 % im oberen Respirationstrakt.
Prg.: Gewöhnlich keine Generalisation.
Monoklonale Gammopathie unklarer Signifikanz (MGUS)
smouldering plasmocytoma
Ig-Erhöhung über Jahre hinweg.
Prg.: In 10 % nach Jahren Übergang in ein Plasmozytom oder B-zelliges malignes Lymphom.
Morbus WALDENSTRÖM
Pathologische IgM-Sekretion.
BURKITT-Lymphom
Morphologisch hochmaligne
Ät.: Assoziiert mit EBV.
Typen:
- Endemisch - Südafrika, Kinder/Jungen 5.-7 Lj., Weichteile des Unterkiefers. Ursache: frühe EBV-Infektion, Malaria?
- Sporadisch - Ubiquitär, 2% der NHL, 20.-30 Lj., nodal
- Immundefensiv - Bei HIV, nodal
Mikro: Große, montone, blastenartige Zellen. Monotone, große Kerne. Schmaler Zytoplasmasaum, atypische Mitosen, prominente Nukleolen. Sternhimmelbild durch eingestreute Makrophagen.
IHC:
- B +: CD 20 +, CD 79 a +
- MIB-1: 90 % +
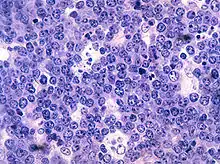 BURKITT-Lymphom, H&E. |
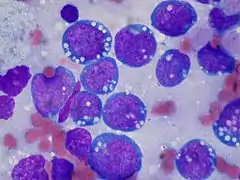 BURKITT-Lymphom, touch prep, Wright stain. |
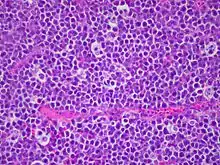 BURKITT-Lymphom, Sternhimmelbild, H&E. |
Extranodales Marginalzonenlymphom des Mukosa-assoziierten Gewebes (MALT-Lymphom)
Niedrig-malignes B-Zell-Lymphom, ausgehend von der Marginalzone von Lymphfollikeln im Bereich von Schleimhäuten/Epithelgewebe außerhalb von Lymphknoten.
Ep.: Selten, Inzidenz 4/100.000/Jahr
Lok.:
- entsteht primär extranodal
- Meist im GIT: Häufigkeitsverteilung Magen:Dünndarm:Dickdarm = 4:2:1
- Speicheldrüsen z.B.: Ohrspeicheldrüse (Glandula parotis)
- Bronchialsystem (BALT)
- Urogenitaltrakt
- Schilddrüse
Ät.:
- Oft auf dem Boden chronisch-entzündlicher Prozesse mit Vermehrung des ortsständigen lymphatischen Gewebes, z.B. bei:
- Chronische Helicobacter pylori Gastritis
- MESA (chronische myoepitheliale Sialadenitis) bzw. SJÖGREN-Syndrom (26,9 % der MESA-Patienten entwickeln ein malignes Non-Hodgkin-Lymphom, davon 88,9 % vom MALT-Typ)
- HASHIMOTO-Thyreoiditis
Zytogenetik: Bestimmte Translokationen und Trisomien.
Histo: Proliferation kleiner lymphoider Zellen mit Expansion der Marginalzone von (reaktiven) Keimzentren. Evtl. Besiedelung der Keimzentren (DD follikuläres Lymphom), Destruktion des umgebenden Gewebes z.B. des Epithels (sog. lymphoepitheliale Läsionen).
IHC:
- B-Marker: CD 20 +, CD 79a +
- CD 3 -, CD 5 - (selten +), CD 10 -, CD 23 -, Cyclin D1 -
- Oft Ig + (IgM, weniger oft IgA, IgG) und light-chain-restriction
DD:
- Benigne reaktive Läsionen: keine Leichtketten-Restriktion
- Mantelzonen-Lymphom: CD 5 +, Cyclin D1 +
- small lymphocytic Lymphoma (CLL): CD 5 +
- Follikuläres Lymphom: CD 10 +
Therapie:
- Antibiose bei Induktion durch chronische Infektion
- Chemotherapie/Strahlentherapie
Prg.: Indolenter Verlauf. Evtl. Übergang in hochmalignes Lymphom.
Lit.:
- Witkowska M, Smolewski P. “Helicobacter pylori Infection, Chronic Inflammation, and Genomic Transformations in Gastric MALT Lymphoma”. Mediators Inflamm., 2013:523170, 2013. DOI:10.1155/2013/523170. PMID 23606792.
- Terada T. “Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) of the ileum in a 35-year-old Japanese woman”. Int J Clin Exp Pathol, 6:951–6, 2013. PMID 23638229.
- Joshi M, Sheikh H, Abbi K, et al.. “Marginal zone lymphoma: old, new, targeted, and epigenetic therapies”. Ther Adv Hematol, 3:275–90, October 2012. DOI:10.1177/2040620712453595. PMID 23616915.
T-Zell-Lymphome (NHL)
T-Zell-Vorläufer-Neoplasien:
- T-Zell-Vorläufer lymphoblastisches Lymphom
Reife/periphere T-Zell-Neoplasien:
- Leukämische/Disseminierte Formen:
- T-lymphozytische Prolymphozytenleukämie
- Chronische granulozytäre Leukämie
- Adultes T-Zell Lymphom/Leukämie
- Kutane Formen:
- Mycosis fungoides
- SÉZARY-Syndrom
- Primär kutanes anaplastisches großzelliges Lymphom
- Lymphomatoide Papulose
- Extranodale Formen:
- Extranodales NK/T-Zell Lymphom, nasaler Typ
- Enteropathie-assoziiertes T-Zell-Lymphom
- Hepatosplenisches T-Zell Lymphom
- Subkutanes pannikulitisches T-Zell Lymphom
- Nodale Formen:
- Angioimmunoblastisches T-Zell Lymphom
- Peripheres T-Zell Lymphom, unspezifiziert
- Großzellig anaplastisches T-Zell Lymphom
Anaplastisches großzelliges T-Zell Lymphom (ALCL)
Ep.: 3 % der Non-Hodgkin Lymphome bei Erwachsenen, 10 - 30 % der großzelligen Lymphome bei Kindern. ALK + ALCL häufiger bei jungen Männern, ALK - ALCL häufiger bei älteren Menschen (leicht erhöhter Frauenanteil).
Lok.: Nodal und extranodal (Haut, Knochen, Weichgewebe, Lunge, Leber, selten zentrales Nervensystem und Darm).
Morph.: Große zytoplasmareiche pleomorphe Zellen, Tumorzellen wachsen kohäsiv in den Lymphknotensinus.
Immunphänotyp: CD 30 +, häufige Expression von zytotoxischen Proteinen, pan-T Zellgene häufig nicht exprimiert; Unterteilung in zwei Gruppen (ALK-positive und ALK-negative ALCL), je nach Vorhandensein einer spezifischen Translokation (Fusionsprotein aus der „anaplasic lymphoma kinase“ (ALK) und verschiedenen anderen Proteinen, häufig Nucleophosmin).
Morph. Varianten:
- Allgemeine Variante
- Lymphohistozytische Variante
- Kleinzellige Variante
Prg.: 5-jährige Überlebensrate bei ALK + 80 %, ALK - 40 %.
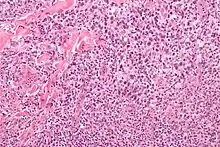 ALCL, H&E. |
 Idem. |
Enteropathie-assoziiertes T-Zell-Lymphom (EATL)
Enteropathy associated T-cell lymphoma (EATL)
Pg.: Kann sich aus einer (refraktären) Sprue entwickeln.
IHC:
- Lymphozyten bei Sprue: CD8 +, CD56 -
- EATL Typ I: CD8 -, CD56 -
- EATL Typ II: CD8 +, CD56 +
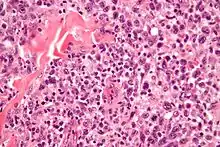
 EATL, H&E. |
Thymus
Thymushypoplasie/-aplasie
- Physiologisch: Altersinvolution - Mikro: Lipomatöser Ersatz
- Erworben: Schwere körperliche Belastung/Krankheitszustände, Mangelernährung, Bestrahlung, Chemotherapie, Kortikosteroide
- Angeboren:
- DIGEORGE-Syndrom (DGS) - Dysmorphie, Herzfehler, Thymushypoplasie, Nebenschilddrüsenhypoplasie (Hypokalzämie) (OMIM)
- severe combined immunodeficiency (SCID) - Thymus- und Tonsillenaplasie
Thymom
Assoziiert mit Myasthenia gravis.
.JPG.webp) Nichtinvasives Thymom Typ B1, Resektat, H&E. |
.JPG.webp) Idem. |
_CK_CAM5-2.JPG.webp) Idem, Immunhistochemie auf Zytokeratin CAM 5.2. |
Milz
Nebenmilzen
Normalbefund.
SF: Intrapankreatische accessorische Milz - Gelegentlich im Pankreas gelegen (klinisch DD Tumor).
Klinik: Asymptomatisch.
Therapie: Keine. Bei Splenektomie zur Behandlung von Bluterkrankungen müssen auch Nebenmilzen mitentfernt werden, um den Therapieerfolg nicht zu gefähren.
Asplenie
Ät.:
- Selten angeboren, z.B. im Rahmen einer Heterotaxie.
- Funktionelle Asplenie mit kleiner vernarbter Milz durch multiple Infarkte bei Sichelzellenanämie.
- Z.n. Splenektomie.
Blutausstrich:
- HOWELL-JOLLY-Körperchen - Kleine runde meist einzeln und randlich liegende basophile Einschlüsse aus DNA-Resten in den Erythrozyten. Vorkommen auch bei gesteigerter Erythropoese und megaloblastärer Anämie.
- Targetzellen (Schießscheiben-Erythrozyten). Vorkommen auch bei Lebererkrankungen, Eisenmangelanämie.
- Siderozyten - Erythrozyten mit feinkörnigen eisenhaltigen Hämosiderin-Einschlüssen. Vorkommen auch bei schweren Anämien.
- Akanthozyten - Erythrozyten mit irregulären hornartigen Ausstülpungen. Vorkommen auch bei Lebererkrankungen und Bluterkrankungen.
Blutbild: Thrombozytose, Leukozytose, Monozytose.
Kompl.: Erhöhte Anfälligkeit für Infektionen mit bekapselten Erregern (Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis)! -> Impfung!
Splenomegalie
Vergrößerte Milz. Norm: Gewicht bis 150 g, Größe 11 x 7 x 4 cm (Merke: 4711).
Vorkommen:
- Hämatologische Erkrankungen (Leukämie, Lymphome, chronische Hämolyse)
- Chronische Entzündungen.
- Infektiöse Mononukleose.
- Pfortaderhochdruck.
Klinik:
- Symptome der Grunderkrankung
- Hyperplenismus -> verstärkter Abbau von Blutzellen -> Zytopenie (Anämie, Thrombopenie und/oder Leukopenie) -> reaktiv hyperplastisches Knochenmark
GAMNA-GANDY-Knötchen
Ät.: Milzstauung z.B. bei Rechtsherzinsuffizienz oder Leberzirrhose, Sichelzellanämie, Hämosiderose.
Pg.: Eisenablagerung (Siderose).
Makro: Kleine rost-braune Knötchen.
Mikro: Ablagerung von Eisen- und Kalzium-Salzen, Bindegewebsvermehrung.
Milzruptur
Ät.: Stumpfes Bauchtrauma, erhöhte Gefährdung bei infektiöser Mononukleose.
Makro: Rißartiger oder flächenhafter Kapseldefekt, Hämatomanteile. Parenchymruptur.
Mikro: Kapseldefekt. Parenchymdestruktion mit Blutungen.
Kompl.: Hämorrhagischer Schock.
Milzinfarkte
Ät.: Z.B. bei Sichelzellenanämie
Makro: Graugelbe dreieckige bis landkartenartige Areale.
Milz bei infektiös-septischem Geschehen
Makro (autoptisch): Aufgelockerte bis zerfließliche Milz, schlaffe Milzkapsel.
Unterteilung:
- infektiös-toxisch (SIRS)
- septisch - Schwere Allgemeinsymptome (hohes Fieber oder Hypothermie, Hypotonie, septischer Schock), Procalcitonin > 2 ng/ml, positive Blutkultur.
„Zuckergußmilz“
Syn.: Kapselhyalinose, Perisplenitis (fibro)cartilaginea.
Ät.: Unklar. Bei vergrößerter Milz (Pfortaderhochdruck etc.).
Schinkenmilz
Bei Amyloidose.
Makro: Das Gewebe ist verfestigt mit glasiger (wachsartiger) Schnittfläche.
Tumoren
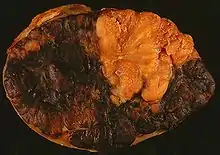
„Bauernwurstmilz“
Noduläre Infiltration der Milz bei Morbus HODGKIN.
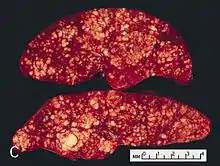 Massive Infiltration der Milz bei Morbus HODGKIN. |
Follikuläres Lymphom
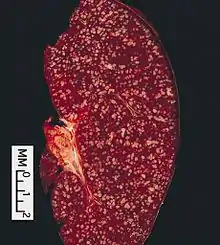 Typischer Aspekt einer Milzbeteiligung bei einem follikulären Lymphom. |
Hamartome
Benigne, ausdifferenzierte Gewebsneubildungen an der "falschen Stelle".
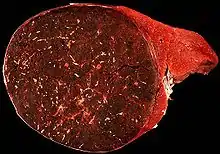 Hamartom der Milz. |
Vaskuläre Läsionen
Hämangioperizytom
Hämangioendotheliom
Peliose
Littoralzellangiom
Ursprung: Littoraltezzeln (Sinuszellen der roten Pulpa).
Makro: Splenomegalie. Knotige, teils auch (pseudo)zystische Schnittflächen.
Histo: Anastomosierende endotheltragende Gefäßkanäle, evtl. pseudopapillär, evtl. kavernös aufgebaut. Plumpe Zellen im Lumen. Evtl. extramedulläre Blutbildung, Siderose und/oder Verkalkungen.
IHC: F VIII +, CD68 +, Lysozym +.
Klinik: Splenomegalie, Anämie, Thrombozytopenie, Fieber, Schwäche, Schmerzen.
Verhalten: Eher benigne. Assoziiert mit verschiedenen Tumoren anderer Lokalisation.
Th.: Splenektomie.
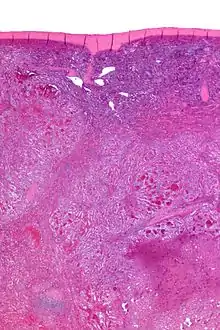 Littoralzellangiom, H&E. |
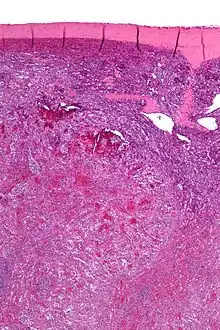 Idem. |
 Idem. |
Literatur:
- Abbott RM, Levy AD, Aguilera NS, Gorospe L, Thompson WM. “From the archives of the AFIP: primary vascular neoplasms of the spleen: radiologic-pathologic correlation”. Radiographics, 24:1137–63, 2004. DOI:10.1148/rg.244045006. PMID 15256634.
- Sauer J, Treichel U, Köhler HH, Schunk K, Junginger T. “[Littoral-cell angioma--a rare differential diagnosis on splenic tumors”]. Dtsch. Med. Wochenschr., 124:624–8, May 1999. DOI:10.1055/s-2007-1024377. PMID 10370385.
- Harmon RL, Cerruto CA, Scheckner A. “Littoral cell angioma: a case report and review”. Curr Surg, 63:345–50, 2006. DOI:10.1016/j.cursur.2006.06.011. PMID 16971207.
- Falk S, Stutte HJ, Frizzera G. “Littoral cell angioma. A novel splenic vascular lesion demonstrating histiocytic differentiation”. Am. J. Surg. Pathol., 15:1023–33, November 1991. PMID 1928554.
- Bisceglia M, Sickel JZ, Giangaspero F, Gomes V, Amini M, Michal M. “Littoral cell angioma of the spleen: an additional report of four cases with emphasis on the association with visceral organ cancers”. Tumori, 84:595–9, 1998. PMID 9862523.
- Dascalescu CM, Wendum D, Gorin NC. “Littoral-cell angioma as a cause of splenomegaly”. N. Engl. J. Med., 345:772–3, September 2001. DOI:10.1056/NEJM200109063451016. PMID 11547761.
Hämangiosarkom
Metastasen in die Lymphknoten
Bei allen malignen Tumoren werden die regionären Lymphknoten präpariert und mituntersucht -> „pN“ im TNM-Staging. Relevant ist die Anzahl und die Lokalisation der befallen Lymphknoten. Extraregionärer Lymphknotenbefall wird meist als Fernmetastase eingeordnet („pM“).
Makro: Die Lymphknoten sind meist verhärtet, vergrößert und zeigen eine grauweiße Schnittfläche.
Mikro:
- Zwei Komponenten:
- Tumorzellen und desmoplastische Stromareaktion (reaktive Bindegewebsvermehrung)
- Lymphatisches Gewebe
- Kapseldurchbruch: ja/nein (strahlentherapeutisch relevant)
DD.: Sinushistiozytose, retikulohistiozytäre Reaktion
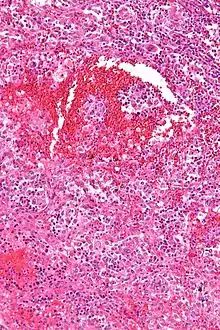
 Lymphknotenmetastase eines malignen Melanoms. |
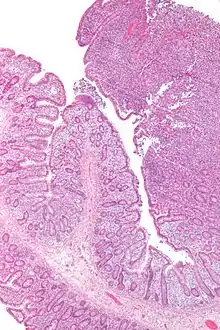
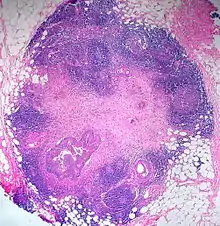 Lymphknotenmetastase eines Colonkarzinoms im paracolonischen Fettgewebe. Atypische glanduläre Elemente mit umgebender Desmoplasie. Im Randbereich Reste des vorbestehenden lymphatischen Gewebes. H&E. |
Lymphgefäße
Malformationen
Kongenitale diffuse pulmonale Lymphangiektasie (PL)
Ät.:
- Primär. Meist sporadisch.
- Sekundär bei Herzfehlern.
Makro: Seröse Pleuraergüsse, evtl. konsekutive Lungenhypoplasie.
Histo: Abnorm erweiterte pulmonale Lymphgefäße (v.a. subpleural und septal).
IHC: D2-40 zur Darstellung der Lymphgefäße.
Klinik: Pleuraergüsse in utero, neonatal akute respiratorische Insuffizienz.
Th.: Intrauterine/postnatale Pleuradrainage, Oxygenierung, ggf. Intensivtherapie.
Prg.: Hohe Letalität.
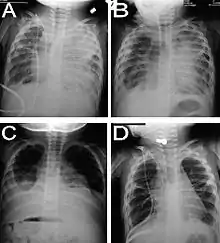 Röntgenthoraxaufnahmen bei PL. |
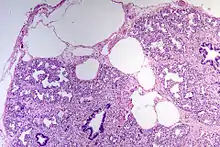 Histologie. |
Literatur:
- Bellini C, Boccardo F, Campisi C, Bonioli E. “Congenital pulmonary lymphangiectasia”. Orphanet J Rare Dis, 1:43, 2006. DOI:10.1186/1750-1172-1-43. PMID 17074089.
- Wilson RD, Pawel B, Bebbington M, et al.. “Congenital pulmonary lymphangiectasis sequence: a rare, heterogeneous, and lethal etiology for prenatal pleural effusion”. Prenat. Diagn., 26:1058–61, November 2006. DOI:10.1002/pd.1555. PMID 16941717.
- Bellini C, Boccardo F, Campisi C, et al.. “Pulmonary lymphangiectasia”. Lymphology, 38:111–21, September 2005. PMID 16353488.
- Esther CR, Barker PM. “Pulmonary lymphangiectasia: diagnosis and clinical course”. Pediatr. Pulmonol., 38:308–13, October 2004. DOI:10.1002/ppul.20100. PMID 15334508.
Weblinks:
Lymphangitis
Ät.: z.B. bakterielle Infektion
Makro: Roter derber Strang in Lymphabflußrichtung. Distal davon gelegener Entzündungsherd.
Primäres Lymphödem
Lymphstau bei Obliteration der drainierenden Lymphwege (inklusive der eingeschalteten Lymphknoten).
Ät.: Z.B. Filariasis, Z.n. Lymphknotenextirpation oder - bestrahlung, Z.n. destruierender Lymphadenitis.
Klinik: Schmerzlose Schwellung, mangelhafte Rückbildung bei Hochlagerung, oft einseitig, STEMMER-Zeichen positiv (Mitbeteiligung der Zehen, dadurch tief einschneidende Falte), v.a. beim chronischen Ödem nicht wegdrückbar (proteinreiches Transsudat, Fibrose).
DD.: Kardiale Ödeme (v.a. untere Extremität, symmetrisch, wegdrückbar), Myxödem bei Schilddrüsenerkrankungen (teigartig, Bein-, Arm- Lidödeme), hydrostatisches Ödem bei Venenerkrankungen (untere Extremität, STEMMER-Zeichen negativ, wegdrückbar), Thrombose (einseitig, livide Schwellung mit Überwärmung, wegdrückbar).
Lymphangiom
Lok.: Z.B. Darmschleimhaut.
Makro: Kleine gelbliche weiche geringgradig erhabene Schleimhautveränderungen.
Histo: Konglomerat aus dilatierten dünnwandigen Lymphgefäßen, ausgekleidet von einem flachen einschichtigen Epithel. In den Lumina feingranuläres eosinophiles Material und Lymphozyten.
IHC: D2-40 markiert das Endothel der Lymphgefäße.
DD.: Lipom.
%252C_HE.JPG.webp) Lymphangiom, H&E. |
Buch-Navigation